MOLEonline documentation
Method section describes the channel detection algorithm used in MOLE
Examples section shows possible uses of MOLEonline application
Tour section guide through MOLEonline application
FAQ Frequently Asked Questions
Updates - list of new features implemented in individual MOLEonline versions
Channels
Channels (tunnels and pores) are highly important structural pathways within proteins and other biomacromolecules. Tunnels connect internal spaces of biomacromolecules with exterior enabling, e.g., substrate/product transport towards enzymes’ active sites, nascent synthesized proteins to leave ribosomal proteosynthetic center via ribosomal exit tunnel, etc. Pores are channels passing through the whole biomacromolecular structure, typically facilitating transport of ions or molecules through cellular biomembranes. (Figure 1)
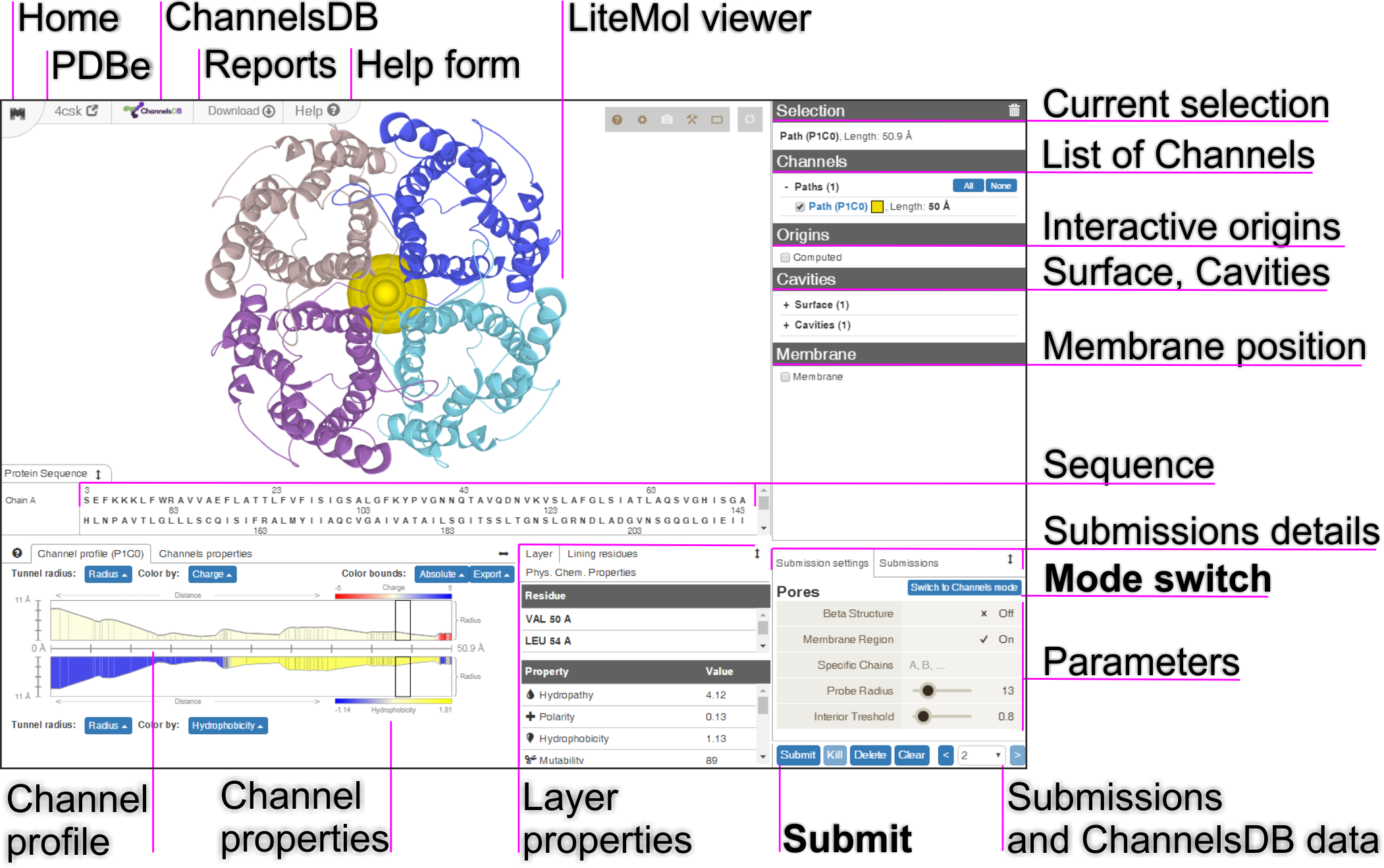
MOLEonline Features
-
Channel calculation
- Tunnels from the stating point to surface or any other end point
- Automatic transmembrane Pores
- Help form with integrated submission for easier support
-
Input
- PDB and mmCIF parsing
- PDBe integration
- PatternQuery language support (ignore residues, start points)
-
Parameters of calculation
- Automatic starting points from cavities in 3D structure - Origin points
- Selection of starting/end points in sequence, residue selection, or XYZ point
- Selection of end points (It is possible to compute tunnels to only 1 user specified exit)
- Several weight functions for computing tunnels (Length, Voronoi scale weight functions)
- Computation of "User Pores" now works for any cavity, not just the "surface".
- Selection of end points with Ctrl+left click
-
Pore mode
- Enhancement of transmembrane pore calculations with use of OPM database or MEMEMBED program
- Vizualization of membrane region
-
Visualization
- LiteMol Viewer
- Layered Channel profile with physicochemical properties and residue lists
- Multiple layers can be selected/deselected to show average properties and list of residues only around important area of the channel.
- Connectivity to ChannelsDB
-
Properties of channels
-
Geometrical properties
- Length, Radius, Free Radius and BRadius
-
Physicochemical properties
- Charge, Mutability, Polarity, Hydropathy, Hydrophobicity, Lipophilicity, Water solubility
-
Geometrical properties
-
Export of results
- Properties of channels - CSV, XML, JSON
- Channel profile in PNG or SVG format
- Output visualization to PyMOL, VMD, and Chimera.
- PDF report or within ZIP archive
- User contributions to ChannelsDB annotations
The calculation setup is realized via an interactive interface consisting of a few components (Figure 1). Structure visualization is achieved by LiteMol Viewer JavaScript application which is capable to visualize the uploaded structure and the computed channels.
Initialization
- Upload from PDBe database via PDBID with Assembly ID (optional) or as a biological unit (recommended).
- Upload PDB or mmCIF formats.
Mode selection and Channel calculation parameters
-
Pore mode - transmembrane pores
- membrane position from Orientations of Proteins in Membranes (OPM) database or calculated with MEMEMBED program.
- Membrane region parameter - calculate pore in transmembrane region only
- Beta structure parameter is recommended for transmembrane regions formed by β-barrel structure
-
Channels mode - identification of channels and other types of pores
-
Start and end points user definition.
- list of residues (selectable from sequence or 3D structure)
- XYZ coordinates
- catalytic site residues from Catalytic Site Atlas (CSA)
- from cofactors
- using PatternQuery language
- on surface using Ctrl + left mouse click (Figure 2)
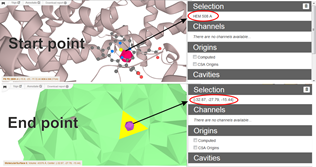
Figure 2 – Interactive selection of start and end points via residues or XYZ coordinates. End point can be selected as a facet on the surface with Ctrl + left mouse click. - Automatic Start points definition in the deepest points within cavities - Origins
-
Cavity parameters
- Probe Radius (default 5 Å) - define coarseness of the surface (larger values are recommended for the detection of larger channels, e.g. 20 Å for detection of peptide channel in ribosome)
- Interior Threshold (default 1.1 Å) - define minimal size of tetrahedrons (smaller allows detection of channels with smaller bottlenecks)
-
Channel parameters
- Origin Radius (default 5 Å) - optimization of starting point within sphere
- Surface Cover Radius (default 10 Å) - only channels with endpoints further than this parameter apart are shown
-
Weight Function selection of priority among channels
- Voronoi scale (default) - adds to original Length and Radius weight function also the differences between the geometrical centers of tetrahedrons and resulting channel spheres. As such Voronoi scale weight function prefers inward facing tetrahedrons. Weight is calculated as W = (L/(D*D+ε))*(S(a)+S(b)), where L is length of edge, D is distance to closest vdW sphere, ε is small number to avoid division by zero and S(a) and S(b) is the distance between center of geometrical centers of two closest tetrahedrons and resulting channel spheres.
- Length+Radius (original in previous MOLEonline version) - weight of edges for channels are calculated as W = L/(D*D+ε),
- Length - weight is calculated just as W = L. This function does not take into account bottlenecks.
- Merge Pores (disabled on default) - merge tunnels starting from the same starting point to form pores
- Automatic Pores (disabled on default) - compute pores in all cavities
-
Channel Advanced parameters
- Bottleneck Radius (default 1.2 Å) - minimum radius of the channel
- Bottleneck Tolerance (default 3 Å) - maximum length of channel segment narrower than bottleneck radius. It can be used for closed channel detection.
- Max Tunnel Similarity (default 0.7) - limit of similarity of tetrahedrons between channels. Longer similar channel is discarded.
-
Start and end points user definition.
Results visualization and analysis
Calculated channels are sorted according to their type to
- Tunnels (from starting point to surface end point)
- Pores (from surface to surface through structure)
- and Paths (between two defined points)
Channels are further sorted according to their cavities by their lengths.
Interactive channels visualization runs in LiteMol Viewer.
Channel profile can be seen in the bottom left corner of the webpage. Two profiles can be visualized simultaneously showing physicochemical properties along channel length (on x-axis):
-
by color filling
- charge - charged amino acid residues (positive ARG, LYS, HIS; negative ASP, GLU).
- hydrophobicity - Kyte and Doolittle scale (ARG -4.5 ; LEU +4.5),
- hydropathy - Cid scale (GLU -1.14; ILE +1.81),
- polarity - Zimmerman scale (ALA, GLY 0; ARG +52)
- lipophilicity - logP-scale - (calculated as octanol/water partition coefficients of Cβ fragments of side-chains (ASN -1.03; TRP 2.59) and mainchain (-0.86) via www.chemicalize.org
- lipophilicity - logD-scale - (calculated as octanol/water distribution coefficients of Cβ fragments of side-chains at pH 7.4 (ASP -3.00; TRP 2.59) and mainchain (-0.86) via www.chemicalize.org
- solubility - logS-scale - (calculated as water solubility of Cβ fragments of side-chains at pH 7.4 (TRP -2.48; ASP 2.63) and mainchain (0.81) via www.chemicalize.org
- mutability - Jones scale (TRP 25; SER 117)
-
by y-axis value
- Radius - radius of maximum sphere in this layer
- Free Radius - channel radius to only main chains of the lining amino acids. May represent maximal radius given by the position of protein backbone, which can be achieved by reconformation of the amino acid side chain aside of the channel or virtual glycine mutation
- BRadius - adds to the Radius value RMSF of the residues along the channel (calculated from B-factors stored with the structure) to reflect local flexibility.
Overall results for all considered channels are also summarized in Channels properties.
Submissions
- Each job has random job ID to ensure private access.
- History of individual submissions within job ID can be browsed in the bottom right corner.
- The user can compare own results with the annotated channels deposited in ChannelsDB if available.
- The user can also send the results as an input of channel annotation to ChannelsDB using a simple form available on top of the page.
Export of results
Results can be downloaded in various formats:
- macromolecule itself in mmCIF format
- channel coordinates in PDB format
- PyMol, VMD, and Chimera visualization scripts
- all information as rich JSON or ZIP (containing CSV, XML) files for further textual analysis
- Channel profiles in PNG or SVG format
- Report for individual channels can be downloaded in PDF format.