FAQ
General
Q: Do you have a question?
- Please don’t hesitate and ask! You can reach us on facebook or email. Contacts are available in the About tab.
- You can also use Help button in your session that will directly point us to your problem at hand.
Q: I am a first time user. What do I do to find channels in my structure?
- First, at least briefly skim through the documentation, as this will give you a better idea who is this service meant for and how you can take the best of it. First, try automatic detection of starting points (just press submit). This might give you a clue, where there are channels in your structure and then you can select those closest to the site of your interest (i.e. active site, cofactors, etc.) or try to select end points of pore by clicking on the molecular surface.. Alternatively, load any existing channels deposited in the ChannelsDB.
Tunnel detection
Q: What should the user do to overcome the program's limitations?
Here we provide several tips how some of the limitations of the implemented algorithms could be overcome as well.
The first limitation is that tunnel is shown only as balls of maximum size along centerline. This limitation can be overcome when starting points are put along the channel to found whether there are bulges along the central line. However the centerline provide good measure to actually show where the channels are localized in the structure.
The second limitation is in use of atom-centered Voronoi mesh. It is difficult to overcome and the real improvement is debatable. The atom centered Voronoi mesh can be replaced e.g. by power diagram, which will lead to some improvement in precision. However, such improvement is small compared to the uncertainties associated with the chosen structures (e.g. X-ray structures with finite resolution, which is generally higher than 0.8 Å).
However, we are certain that a user can face many problems trying to analyze his/her specific molecule. We are ready to help users and we provide a feedback via e-mail or Facebook page to resolve specific problems.
Q: I have tried to find tunnels, but I was not able to get any, or they do not seem to be relevant to my query, what should I do?
This can be caused by a number of reasons. Always bear in mind, that we are happy to help you with any issues you might come across, so don’t be shy and ask for a help! Here is a list of the most common issues:
- Wrong setup of ProbeRadius, InteriorThreshold or filtering criteria.
- Wrong location of the active site (channel starting point).
- Substrate is blocking the channel.
- There is no channel at all.
-
Setting up the two key parameters can be at times indeed critical for proper identification of channels. These parameters can be think of as a lower bound (InteriorThreshold) and an upper bound (ProbeRadius) threshold for a channel radius. Therefore, if the expected channel is supposed to be wide in its radius the ProbeRadius parameter needs to be increased preferably above the expected channel radius. Likewise if the channel is expected to pass through a constringency region, which features a local narrowing, the InteriorThreshold needs to be set below the expected minimal channel radius.
Finally, all the channels are a subject of filtering step. Try adjusting the values of MaxTunnelSimilarity, BottleneckRadius or BottleneckLength. It is always good practice to set BottleneckRadius a little lower than InteriorThreshold and allow BottleneckLength to cover for small local narrowings beyond this threshold.
Example:
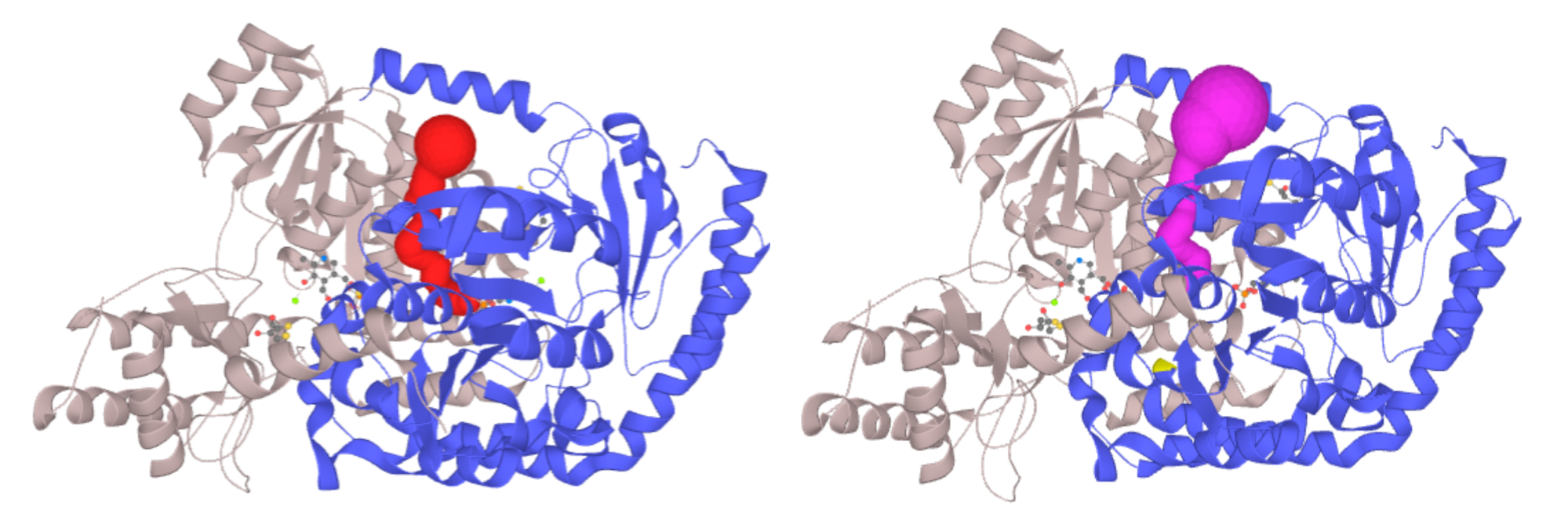
Left - Channel of PLP-dependent Acetyl-CoA transferase CqsA (PDB ID: 3KKI) ProbeRadius set to value 5. Right - ProbeRadius set to 20 showed overestimation of the parameter. -
In certain cases even if the two major parameters are set correctly the deemed channels still cannot be seen. The MOLE algorithm is designed to identify channels leading to the buried sites inside the protein structure, therefore, if your active site is located in a shallow pocket or even on the protein surface, the channel analysis will not help here.
Alternatively, if your active site is indeed buried within a protein body, you can enlarge the space to be searched for potential channel starts by increasing OriginRadius parameter.
Example:
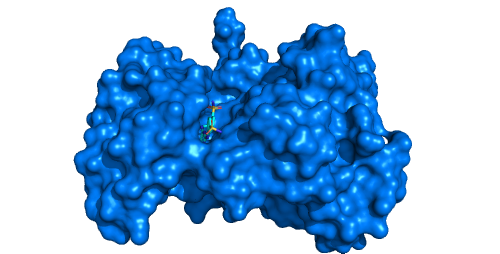
Tyrosine kinase EphB4 (pdb id: 2vwx) with shallow open pocket, for which the channel analysis is pointless. -
MOLE in its default settings considers only protein/DNA/RNA atoms, effectively discarding any het groups and solvent molecules prior to the calculation. However, even this can be modified in order to keep for instance cofactors in the structure prior to the calculation. The algorithm heavily relies on the correct annotation in the input structure. That is the annotation of the _atom_site.group_PDB field, which is set to either ATOM or HETATM in the mmcif/PDB language.
Example:
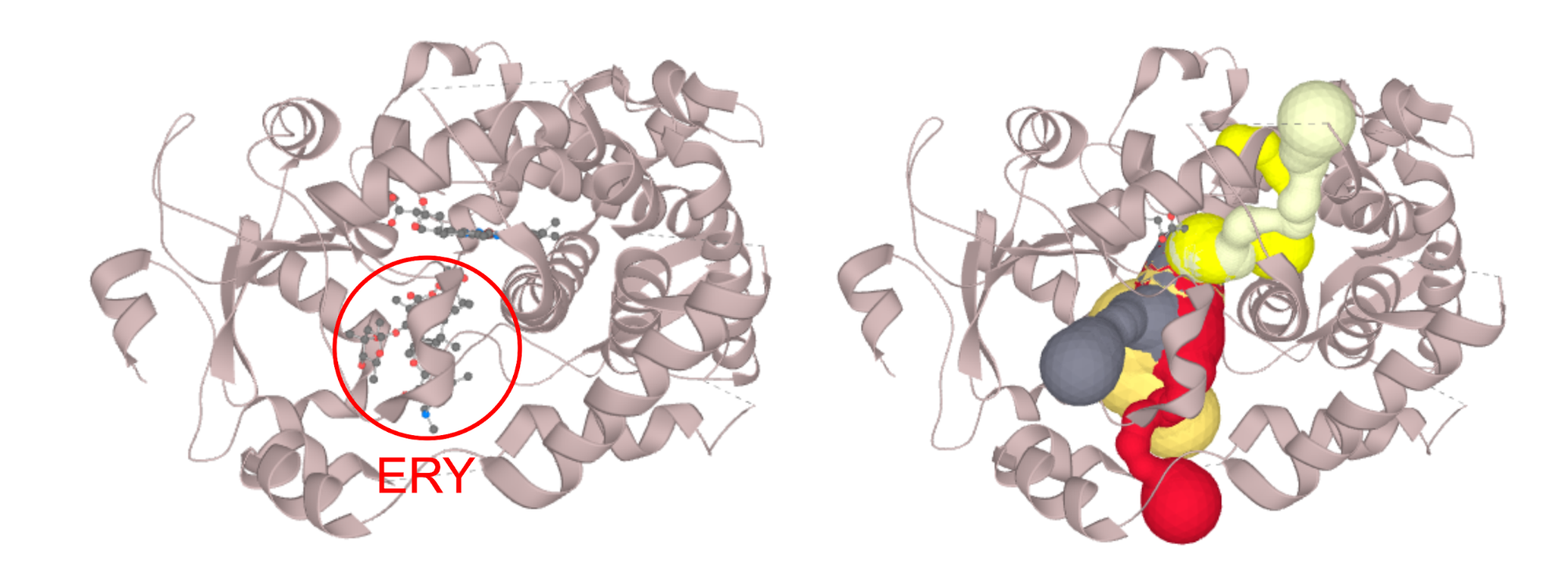
Structure of CYP3A4 with bound erythromycin (PDB ID: 2J0D; left) which is blocking the opening of the access channels. Discarding of the erythromycin molecule allows the proper calculation of access channels (right). In case you require to switch off a certain part of the structure (residues, or even certain atom groups) prior to the calculation. You can select those in the active atoms/residues section. We strongly encourage you to try the PatternQuery syntax, as this is the most flexible option. For example to discard all the alanine residues you just type in: Residues('ALA'). To discard ammonium group of lysine residues capable of carrying protons simply add: AtomNames('NZ').Inside(Residues('LYS')). If you want to combine both, just join them to: Or(Residues('ALA'), AtomNames('NZ').Inside(Residues('LYS'))). More details on PatternQuery syntax can be found in the documentation.
- The final option, if none of the above helped. There is always a possibility that there are no reasonably sized channels in the protein conformation you provided. Drop us an e-mail, so we can take a look, whether or not this is the case!
Q: I cannot detect my transmembrane pore. Or the result is unlikely to be of a biological relevance.
Give it a go in the pore mode. This is the most comfortable option for the majority of transmembrane molecular pores, as it requires almost no setup. After the calculation check the position of membrane. If the position is not of a biological relevance the pore will not be found or will be likely wrong. If the pore module fails, please let us know. There is always a room for improvement.
In most of the transmembrane channel structures the putative centerline can be seen by a naked eye. In this case switch back to the channels mode, display the cavity from the upper right panel and select pore end points with the Ctrl + left click option. Lastly, identify them to be channel end points in the menu. Do not forget to change parameters of ProbeRadius and InteriorThreshold, as those are different in pore and channels mode.
Q: My channel is visibly present and MOLEonline do not detect it. Why?
If the channel is large enough to be visible by naked eye, it can be larger usually shown as a part of the surface and consequently, it is necessary to enlarge Probe radius to cover such structure.
Q: My channel has probably too small radius to be detected or not?
When the channel is too narrow in a certain part, it sometimes helps to lower Interior Threshold parameter.
Physicochemical Properties
Q: It is unclear whether reporting the averages of some channel properties will be that useful?
MOLEonline enables analysis of some basic physicochemical properties - hydropathy, hydrophobicity, polarity, lipophilicity, solubility and mutability, lipophilicity (logP and logD), water solubility (logS-like scale of channel-lining residues). We believe that this is a step forward in channel analysis, but we are certain that it will take some time to optimize analysis of these features and to fully understand their biological relevance. Considering all pros and contras, we decided to keep this functionality in the current version to enable “experiments” with this feature to a broad scientific community.
Q: Regarding relative mutability: Isn't any residue mutatable?
Every amino acid can be mutated; however some amino acids can be mutated more safely with respect to structure/stability/folding of a protein. The relative mutability index reflects this feature, e.g. Pro and Trp residues are typically structurally important and their mutations often lead to unstable proteins or proteins with alternated structures. The relative mutability index which we employ is based on the analysis of the protein sequences (similarly as well known BLOSUM and PAM matrices used in sequence alignments) and it was taken from Jones, D.T., Taylor, W.R. and Thornton, J.M. (1992) The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci., 8, 275-282.